Общее описание заболевания
Нормальная свертываемость крови предотвращает и останавливает кровоизлияния в мышцы и суставы (гемартрозы и гематомы), а также кровотечения при порезах и царапинах, которые могут возникнуть при активной повседневной жизни любого человека.
История гемофилии
В 12 веке Абу аль Касим, врач служивший при дворе одного из арабских правителей Испании, первым описал симптомы гемофилии. Он писал о нескольких семьях, в которых дети мужского пола умирали от небольших повреждений.
Современное научное исследование гемофилии ведется с XIX века. Впервые термин «гемофилия» был введен в 1828 году швейцарским врачом Хопфом.
Первой высокопоставленной носительницей гемофилии считается английская королева Виктория. В «наследство» от нее это заболевание было получено царствующими семьями Германии, Испании и России.
Проявления гемофилии
Чем тяжелее гемофилия, тем раньше проявляются признаки кровоточивости. Неизбежные падения и ушибы, которые случаются у ребенка, начинающего ходить, могут вызвать синяки на коже и кровотечения из слизистых оболочек губ и языка.
В возрасте 1-3 лет могут начаться поражения мышц и суставов, с болезненными припухлостями, ограничением движений рук и ног. Обширные гематомы, бывающие опасными, могут вызываться внутримышечными инъекциями.
При болезни Виллебранда наиболее частыми симптомами бывают кровотечения из слизистых носа.
Диагностика гемофилии
Иногда наличие диагноза вызывает сомнения, даже при повышенной кровоточивости у самого больного или наличии у других членов семьи гемофилии. Точная диагностика может быть сделана только измерением уровня соответствующего фактора свертывания крови. Такие анализы проводятся в специализированных лабораториях гематологических центров.
Наследование гемофилии
Гемофилия А и В поражает почти исключительно мужчин, а передается по женской линии. По статистике ВОЗ примерно 1 младенец мужского пола из 5000 рождается с гемофилией А, вне зависимости от национальной или расовой принадлежности. Приблизительно одна треть больных гемофилией А не имела у предшествующих поколений подобных расстройств.
Наследование болезни Виллебранда и гипопроконвертинемии не сцеплено с полом.

нажмите, чтобы увеличить
Тяжесть гемофилии
Гемофилию различают по форме тяжести, обусловленной уровнем фактора свертывания крови.
Гемофилия тяжелой формы. Уровень FVIII или FIX менее 1%. Кровоизлияния в суставы, мышцы и другие органы случаются при минимальных или даже незаметных повреждениях.
Гемофилия средней формы тяжести. Уровень FVIII или FIX 1-5%. Кровотечения возникают из-за явных незначительных повреждений, а также после различных операций и удалений зубов.
Гемофилия легкой формы. Уровень фактора FVIII и FIX 6-30%. Кровоизлияния обычно следуют за крупными повреждениями, хирургическими операциями или удалением зубов. Эта форма может не быть диагностирована достаточно долго.
Тяжесть болезни Виллебранда зависит от её типа и подтипа.
Лечение гемофилии
Сейчас гемофилию А и В лечат концентратами соответствующих факторов свертывания крови. Комплекты поставки препарата включают в себя лиофилизированный (высушенный) фактор свертывания, воду для растворения, а также приспособления для внутривенных инъекций. Таким образом, имеется возможность для лечения пациента вне медицинского учреждений. Этот тип лечения называется домашним.
Гематологи с раннего детства при тяжелой форме заболевания рекомендуют больным гемофилией профилактическое лечение. При таком лечении, инъекции фактора производятся не по факту кровоизлияния (кровотечения), а вне зависимости от него, по специальному расписанию, составленному врачом-гематологом.
В последние годы в России удалось достичь уровня обеспечения пациентов с гемофилией А и В на уровне развитых стран Запада. Благодаря этому, новое поколение больных гемофилией минимально инвалидизировано. Препараты проходят вирусную инактивацию и шанс заразиться известными на данный момент вирусными инфекциями, передаваемыми через кровь, в современных условиях стремится к нулю.
Гемофилия
Общие сведения
Гемофилия — что это за болезнь? Гемофилия (латынь – haemophilia) представляет собой генетически наследуемое заболевание, в основе которого наследственный дефицит (снижение активности) плазменных факторов свертывания крови (VIII — гемофилия А или IX — гемофилия В), проявляющееся геморрагическим синдромом (снижением свертываемости крови, выраженной склонностью к кровоизлияния/кровотечениям). Под фактором свертывания крови подразумевается белок, содержащийся в тромбоцитах/плазме крови, который и обеспечивает свёртывание крови.
В норме уровень активности фактора свертывания крови варьирует в пределах 50-150%.
Тип наследования: Х-сцепленный рецессивный и в подавляющем большинстве случаев (около 70%) прослеживается положительный семейный анамнез. Гемофилия обусловлена мутациями в гене FIX (Xq27) или FVIII (Xq28). Реже встречаются случаи без наличия семейного анамнеза на фоне спорадических мутаций (Википедия). Спорадическая генная мутация может произойти в период овуляции/сперматогенеза в любом поколении и на протяжении нескольких поколений женщин-носителей оставаться скрытой, пока несущая ген гемофилии Х-хромосома случайно не передастся мальчику. При этом, в сперматогенезе мутации в гене возникают в 4-5 раз чаще, чем в оогенезе. То есть, матери в 80-85% спорадических случаев заболевания являются носителями генной мутации, которые возникли в зародышевых клетках отца. Прослеживается сильная корреляция между возрастом отца и риском получения дочерью носительницей от него мутации: средний возраст отца 40 лет.
Гемофилия (несворачиваемость крови) — достаточно редкое заболевание (ежегодные показатели заболеваемости в человеческой популяции варьируют в пределах 7-15 случаев/100 000 населения). При этом, на долю гемофилии А (ГА) приходится 78-88% из общего числа случаев заболеваний. В подавляющем числе случаев болеют гемофилией лица мужского пола. Женщины являются кондукторами (носителями/проводниками) гена гемофилии и передают части своих сыновей это заболевание.
Единичные случаи гемофилии встречаются и у лиц женского пола при наследовании гена от больного гемофилией отца и от носителя гена — матери или у женщин с наличием мутации гена на одной хромосоме при неактивном гене на другой хромосоме (болезнь Шерешевского-Тёрнера). Крайне редко могут быть клинические проявления у некоторых женщин – носителей фактора свертывания крови (FIX)/мутации гена фактора свертывания крови (FVIII).
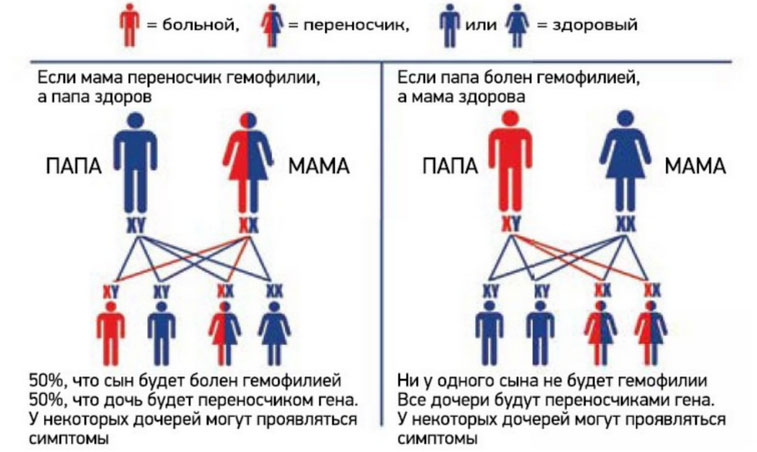
Ниже приведена схема наследования гемофилии, согласно которой вероятность рождения сыновей без признаков или с признаками гемофилии у женщин-носительниц патологического гена и здорового мужчины одинакова (50:50). При этом у некоторых дочерей может присутствовать симптоматика заболевания, а вероятность рождения дочери-переносчика гена составляет 50%. А мужчина больной гемофилией сможет зачать здоровых детей с женщиной не носительницей патологически измененного гена. При этом все рожденные дочери будут переносчиками заболевания.
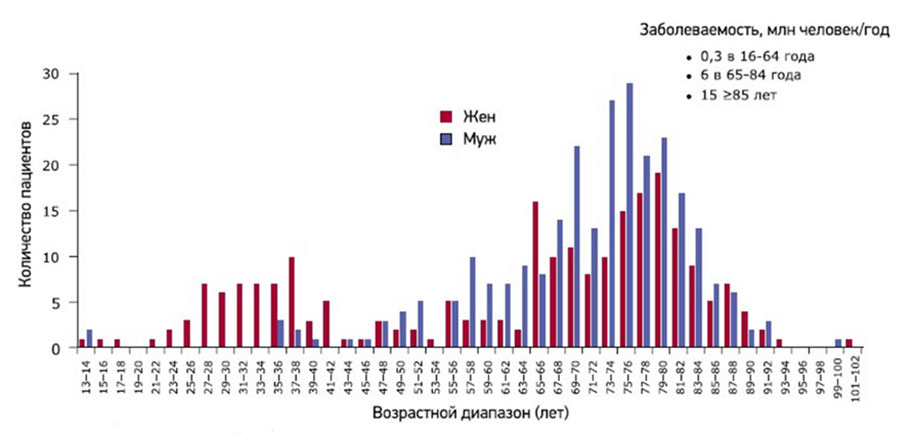
Кроме генетически обусловленных (наследственных) форм выделяют приобретенные формы гемофилии (болезнь Розенталя), в основе которых дефицит фактора ХI и которая относится к группе редких коагулопатий. Приобретенные формы гемофилии встречаются крайне редко и в основе их развития — появление антител к факторам свертывания, что характерно для аутоиммунных/миелопролиферативных заболеваний, гиперчувствительности к лекарственным препаратам, заболеваниям кожи и беременности. Приобретённая гемофилия наиболее распространена в возрастном периоде 69-80 лет, однако среди женщин патология часто встречается в репродуктивном возрасте. В целом распространенность приобретенной гемофилии составляет 1,5 случая/1 млн населения /год (рис. ниже).
 Зачастую в литературе гемофилия А называется болезнь «викторианская» или «царская болезнь», которая появилась в России, благодаря потомкам королевы Виктории и которой страдал сын Николая 2, Алексей. Злосчастный ген унаследовала мать ребенка (Александра Фёдоровна, являвшейся внучкой Виктории), который она передала сыну. Распространению гемофилии среди царствующих лиц Европы способствовали также браки между близкими родственниками, практиковавшиеся с целью укрепления династий и политической целесообразности, примеры которых хорошо известны в истории тех или иных монархий.
Зачастую в литературе гемофилия А называется болезнь «викторианская» или «царская болезнь», которая появилась в России, благодаря потомкам королевы Виктории и которой страдал сын Николая 2, Алексей. Злосчастный ген унаследовала мать ребенка (Александра Фёдоровна, являвшейся внучкой Виктории), который она передала сыну. Распространению гемофилии среди царствующих лиц Европы способствовали также браки между близкими родственниками, практиковавшиеся с целью укрепления династий и политической целесообразности, примеры которых хорошо известны в истории тех или иных монархий.
Патогенез
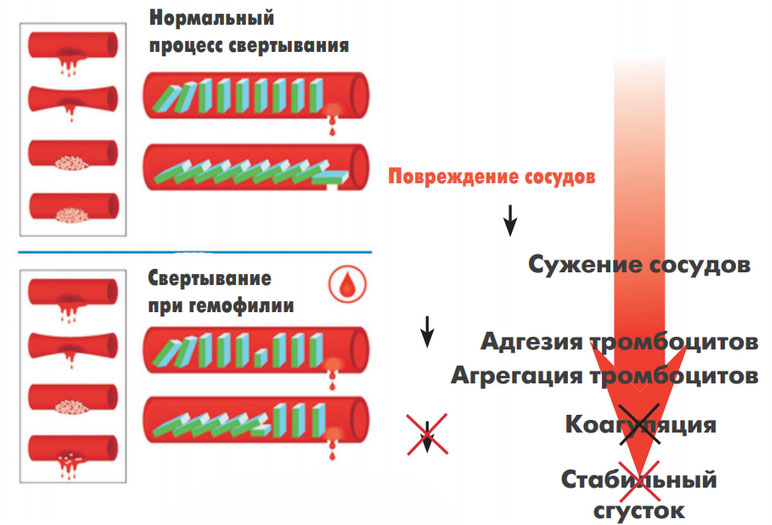
В основе патогенеза гемофилии лежит дефицит плазменных факторов (VIII, IX, XI) свертывания крови, что вызывает нарушение процесса ее свертывания во внутреннем коагуляционном звене гемостаза (образование тромбопластина) и обуславливает гематомный отсроченный тип кровоточивости. Первый этап свертывания крови — это процесс тромбопластинообразования, который нормально протекает лишь при условии достаточной концентрации факторов VIII и IX. Его продолжительность составляет 12-15 минут, а последующий процесс свертывания крови после появления активного тромбопластина в крови происходит практически мгновенно.
Именно нарушение этой плазменной фазы гемостаза обуславливает характерный для гемофилии тип кровоточивости. Кровотечение сразу после травмы может отсутствовать, поскольку первичный (сосудисто-тромбоцитарный) гемостаз функционирует нормально, что и обеспечивает формирование первичного тромба.
Поскольку первичный тромб окончательную остановку кровотечения обеспечить не в состоянии, страдает вторичный гемостаз — формирование фибринового (окончательного) тромба, то кровотечение возобновляется и возникает внезапно через несколько часов (на следующие сутки) после хирургического вмешательства/травмы. При этом, несмотря на его длительность нарастание объема кровопотери отсутствует, что и является его особенностью. Ниже на рисунке схематически приведен патогенез гемофилии.
Классификация
Различают несколько типов заболевания:
Классификация гемофилии А и В — по степени тяжести заболевания, определяемая активностью FVIII и FIX, в соответствии с чем выделяют 3 формы тяжести:
Причины
Непосредственной причиной развития гемофилии А/В является генные мутации Х-хромосомы в зоне длинного (q27-q28) плеча. Около 70-75% больных имеют геморрагический синдром в семейном анамнезе (у родственников), а у 25-30% наследование заболевания в семейном анамнезе не прослеживается, то есть, в Х-хромосоме имеет место спонтанная мутация генов.
Выявлено, что ген, который кодирует процесс синтез фактора VIII, расположен в локусе Xq 28 на длинном плече Х-хромосомы и состоит из 25 интронов/26 экзонов. По размеру ген фактора VIII насчитывает 186 тыс. парных оснований ДНК. При этом мутации в этом гене могут происходить по разному типу: делеции, дупликации, включения новых оснований, сдвиги рамки считывания. В большинстве случаев наблюдается инверсия последовательности нуклеотидов, в частности, инверсии в гене VIII фактора интрона 22, что обуславливает низкий уровень этого фактора, а также вызывает тяжелые фенотипические проявления.
Ген фактора IX расположен на длинном плече X-хромосомы в локусе Xq 27 и включает 7 интронов/8 экзонов. При этом, мутации в гене фактора IX происходят в 7-10 раз реже, чем в гене фактора VIII. При тяжелой форме гемофилии часто происходят два вида генных мутаций.
Симптомы
Степень клинических проявлений и нарушений свертывания крови зависит от уровня активности фактора в крови, но в клинической практике прямая корреляция между клиническим фенотипами заболевания и лабораторными данными существует не всегда. Клинические симптомы гемофилии манифестируют геморрагическим синдромом, проявляющегося в виде гемартрозов, на долю которых приходится 70-80% проявлением гемофилии; гематом и кровотечений. Типичным является частое рецидивирование геморрагического синдрома, а его отличительной особенностью — неадекватность тяжести травмы.
Гемартрозы
Гемартрозы (кровоизлияния в суставы) являются типичным проявлением заболевания, а источником кровотечений в суставе является преимущественно суставная сумка. При отсутствии своевременного адекватного лечения, кровотечение может продолжаться до полного наполнения сустава кровью. У детей раннего возраста кровоизлияния в суставы встречаются редко. Однако, по мере взросления ребенка и повышения двигательной активности, возрастания нагрузки на суставы их частота резко возрастает. Наиболее часто отмечаются гемартрозы коленных (48%), локтевых (24%) и голеностопных (16%) суставов, значительно реже поражаются плечевые/тазобедренные (5-8%) и мелкие суставы стоп/кистей. Случаи поражения суставов позвоночника встречаются крайне редко. Как правило, в зависимости от тяжести заболевания/возраста пациента поражаются несколько суставов (от 1 до 8-10).
Кровоизлияние в суставы развивается преимущественно без видимых причин. Характерным является быстрое увеличение сустава в объеме, локальная гиперемия/гипертермия, резкая болезненность и напряженность кожи над суставом. Ниже на рисунке приведена клиническая симптоматика гемартроза, в зависимости от объема кровоизлияния в полость сустава.
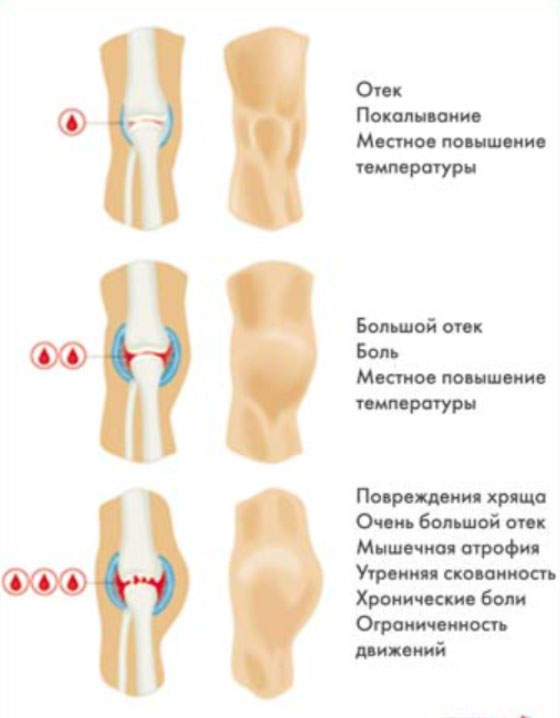 Отмечается нарушение функции сустава, формируется контрактура: движения в пораженном суставе ограничены, сустав деформируется и при распрямлении конечности принимает неправильные формы. После первых кровоизлияний, как правило, кровь в синовиальной полости медленно и постепенно рассасывается, а функция сустава также постепенно восстанавливаются. Однако, если кровоизлияние в определенный сустав происходит часто, то чаще начинается реакция постепенного развития артрита, а такой сустав принято называть название «суставом-мишенью».
Отмечается нарушение функции сустава, формируется контрактура: движения в пораженном суставе ограничены, сустав деформируется и при распрямлении конечности принимает неправильные формы. После первых кровоизлияний, как правило, кровь в синовиальной полости медленно и постепенно рассасывается, а функция сустава также постепенно восстанавливаются. Однако, если кровоизлияние в определенный сустав происходит часто, то чаще начинается реакция постепенного развития артрита, а такой сустав принято называть название «суставом-мишенью».
При частых кровоизлияниях в сустав и отсутствии скорой/адекватной заместительной терапии развивается гемофилическая артропатия, представляющая собой стойкое деформирующее изменения суставов.
Постепенно артроз поражает хрящ и капсулу сустава, а позже патологический процесс распространяется на мягкие ткани/прилегающие кости. Воспаленная синовиальная оболочка постепенно утолщается, а местные сосуды легко повреждаются, что и является источником повторных кровоизлияний. Развивающийся фиброз капсулы/окружающей ткани деформирует сустав и резко ограничивает его подвижность. Под воздействием протеолитических ферментов разрушается хрящ сустава (дегенерация хряща), а в прилегающей кости развивается остеопороз с образованием кист, наполненных студенистой жидкостью.
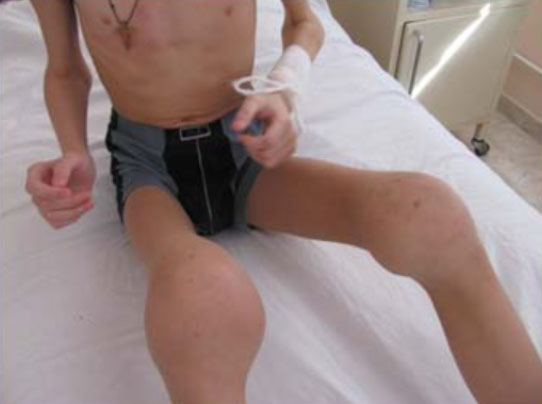
Мускулатура, окружающая сустав, постепенно атрофируется, что увеличивает нагрузку на сустав. При гемофилической артропатии различают следующие формы гемартроза: острый гемартроз (первичный, рецидивирующий); постгеморрагический синовиит (острый, подострый, хронический); деформирующий остеоартроз; анкилоз (фиброзный/костный). Как правило, раннее возникновение гемартрозов способствует устойчивому поражению поверхностей суставов и его компонентов, а развитие множественных артропатий приводит к глубокой инвалидизации пациента (рис. ниже).
Гематомы
Еще к одному характерному проявлению гемофилии относятся гематомы различной локализации (межмышечные, подкожные, поднадкостничные, ретроперитонеальные, субсерозные и др.), для которых характерна выраженная тенденция к распространению. Излившаяся кровь длительный период остается жидкой, что способствует ее легкому проникновению в прилегающие ткани и быстрому распространению вдоль фасций. Процесс рассасывания гематом медленный.
Наибольшую угрозу для жизни пациента представляют ретроперитонеальные гематомы, симптомы которой напоминают клинику острого аппендицита. Причиной частичной непроходимости кишечника могут быть субсерозные гематомы, которые зачастую ингибируют стенку и содержимое прорываться в просвет кишечника. Также, обширные гематомы в ряде случаев могут сдавливать крупные артерии/нервные стволы, вызывая расстройства кровообращения и нарушение чувствительности. Более того, сжатие гематомой кровеносных сосудов может вызывать некротизирование тканей и образование «гемофилических псевдоопухолей».
Клинически процесс формирования гематом может сопровождаться как местными (болезненность, ощущение жара, покалывание/онемение, ограничения движения за счет мышечного спазма), так и общими симптомами (слабость/недомогание, нарушении аппетита/сна лихорадка при рассасывании гематомы). Также, при излитии крови в большом объеме может развиваться анемия.
Кровотечения
У больных гемофилией кровотечения являются легко и часто встречаемыми, они обильны и длительны, могут происходить в любое время суток. Частота кровотечения во многом определяется тяжестью гемофилии. Так, частота эпизодов кровотечений у больного с тяжелой формой гемофилии А может варьировать в пределах (15-35 раз/год и более). У пациентов с среднетяжелой/легкой формой гемофилии А и В кровотечения развиваются значительно реже. Традиционные меры остановки кровотечения эффекта не дают.
Выделяют наружные кровотечения (из слизистых оболочек ротовой полости, десен, носа) встречающиеся в 75% случаев и внутренние кровотечения (почечные, желудочно-кишечные, кровоизлияния в головной мозг/мозговые оболочки), которые встречаются относительно редко и возникают преимущественно после различных инструментальных манипуляций/в результате травм. Длительные почечные кровотечения встречаются у 14-30% пациентов с тяжелой формой гемофилии, при этом, гематурия чаще возникает спонтанно и может привести к развитию анемии. Как правило, развитие внутреннего кровотечения у большинства больных сопровождается острой болью. Выделяют две категории кровотечений, критерием которых служит степень опасности для жизни: жизнеугрожающие и обычные.
К жизнеугрожающим кровотечениям относятся кровотечения в жизненно важные органы. Их частота варьирует в пределах 5-10%, однако они часто являются причиной параличей/летальных исходов (кровоизлияния в спинномозговой канал, мозговые оболочки, головной мозг, в ЖКТ). Особую опасность представляет ретрофарингеальное кровотечение, поскольку за счет скопления в верхних дыхательных путях сгустков крови может спровоцировать удушье. Как правило, тяжесть кровоизлияния не соответствует интенсивности/обширности травмы, а кровоизлияния могут возникать отсрочено (от 6-12 часов до суток) после травмы в зависимости от степени дефицита фактора. Это обусловлено тем фактом, что в основе первичной останови кровотечения лежит сосудисто-тромбоцитарное звено гемостаза, функции которого при гемофилии не нарушены.
Анализы и диагностика
Установление диагноза гемофилия проводится на основе изучения семейно-наследственного анамнеза, физикального обследования (наличие у пациента проявлений кожного геморрагического синдрома в виде множественных гематом/экхимозов различной степени выраженности), клинических проявлений (поражения суставов в виде отека/деформации, локального повышения температуры кожи, нарушения объема движений/подвижности суставов, гипотрофии мышц конечности и др.), а также данных лабораторного коагулологического обследования (активированное частичное тромбопластиновое время, тромбиновое время, протромбиновое время, время свертывания крови, кровиконцентрация фибриногена по Клауссу, снижение уровня фактора VIII/IX и др.). Также может проводится молекулярно-генетическое исследование.
С целью оценки тяжести поражения суставов и состояния органов, в которых отмечаются профузные кровотечения (почки, желудок, кишечник) проводят рентгенологическое исследование, УЗИ/КТ/МРТ суставов/мягких тканей, органов грудной клетки и брюшной полости, мочевыводящих путей, головного мозга, эзофаго-гастро-дуоденоскопию/колоноскопию.
Наиболее часто диагноз гемофилии устанавливается в раннем детском возрасте, когда ребенок начинает ходить и родители отмечают, что у него кровь не сворачивается.
Средний возраст постановки диагноза «гемофилия», тяжелая форма составляет 9 месяцев; при среднетяжелых формах — 22 месяца, а легкие формы заболевания диагностируют у детей в более позднем возрасте, преимущественно после инвазивных вмешательств/удаления зубов. Скрининговым тестом нарушений свертывающей системы является время свертывания и при удлинении этого показателя проводятся исследования уровня факторов в крови, на основании чего и устанавливается диагноз «гемофилии» и определяется тяжесть заболевания.
Лечение
Гемофилия — серьезное и неизлечимое заболевание, но своевременно начатое правильное лечение позволяет избежать инвалидности и преждевременной смерти. Лечение гемофилии заключается в заместительном применении концентратов отсутствующих у больного факторов свертывания. Это могут быть препараты, приготовленные из плазмы доноров (концентрат FVIII, FIX, VIII+vWF), или рекомбинантные концентраты (Адвейт, Когенэйт ФС, Рефакто АФ, Иннонафактор, НовоСэвен, Бенефикс, Нувик, РеФакто АF). Причем предпочтений препаратов той или иной группы нет. Все они вводятся внутривенно капельно. Лечение вне сомнения улучшает состояние больного: предотвращает кровотечения и спонтанные кровоизлияния, останавливает прогрессирование костно-суставных изменений.
Возможны следующие варианты лечения больного:
Профилактическое заместительное лечение концентратами факторов — необходимое условие для больных с тяжелой гемофилией и средней тяжести. Это основное направление у больных с 2 до 17 лет (весь период роста больного). При тяжелом течении такое лечение продолжается долго и зависит от возраста. Профилактическое лечение начинается с раннего детства после первого кровотечения или гемартроза. У взрослых больных чаще всего применяется лечение «по требованию» предупреждение для предупреждения опасных кровотечений.
Профилактическое лечение разрабатывается с учетом особенностей больного: подбирается тип фактора, доза, частота введения, чтобы минимизировать риск кровотечений и продолжительность курса. Первичная профилактика эффективна для предотвращения осложнений со стороны суставов и замедляет прогрессирование заболеваний суставов. Поскольку введение препаратов предполагается частым и постоянным, больным устанавливаются венозные катетеры иногда центральный венозный катетер.
В режиме профилактики внутривенно капельно вводятся:
Профилактическое введение проводят утром для того, чтобы максимальный уровень фактора был днем в период активности больного. Родственники больных обучаются проведению инфузии в домашних условиях и правильному уходу за венами. Поддержание фактора свертывания на уровне 2-5% является достаточным для многих больных и позволяет вести нормальную жизнь. Постоянная заместительная терапия нуждается в лабораторном контроле (определяется активность дефицитного фактора и тест восстановления его).
Использование низкой дозы или несоблюдение режима снижает эффективность лечения и ухудшает состояние. Наоборот, очень высокая доза вызывает развитие тромбозов. Вторичная профилактика бывает ранней (если проводится с 5 лет) и поздней (если проводится с 10 лет). Она замедляет развитие артропатий и обеспечивает качественную деятельность и активность. При различных состояниях (почечное кровотечение, гемартроз, кровоизлияние в мышцы, кровоизлияние в мозг, кишечные кровотечения) важно незамедлительное проведение заместительной терапии — с первого часа после травмы или признаков кровоизлияния, чтобы не допустить значимых кровопотерь.
В лечении легкой степени гемофилии А при различных ситуациях эффект может оказать гемостатическая терапия:
Данные препараты можно применять совместно с заместительной терапией при удалении зуба, лечении ран и даже при проведении операций на паренхиматозных органах.
Перспективным является применение моноклонального антитела эмицизумаб (препарат Гемлибра), который имитирует функции недостающего фактора VIII. Он связывает фактор IXа c X, восполняя отсутствующий фактор. Рассматривается как профилактический препарат для больных гемофилией А с ингибиторами к фактору и при их отсутствии. Вводится подкожно в разных режимах — один раз в неделю, в две или четыре недели. В странах ЕС и России Гемлибра разрешена только для больных гемофилией А ингибиторной формы.
Ситуация при лечении ухудшается, если у больного образуются ингибиторы факторов свертывания, что является тяжелым осложнением, возникающим при лечении. Чаще всего ингибиторы появляются при тяжелой форме гемофилии А (до 30%), а при тяжелой форме гемофилии В они обнаруживаются только у 3-5% больных. Ингибитор чаще всего развивается в первые дни введения фактора (50-100 дней) или после введения большого его количества при операции. Ингибиторы — это антитела, вырабатываемые иммунной системой, которые связываются с введенным фактором и блокируют его действие. В результате этого достаточный уровень фактора не достигается и контролировать кровотечение становится невозможным. Больным приходится повышать дозу и частоту введения, но и это оказывается неэффективным. В связи с этим первые 20 дней нужно определять уровень ингибитора раз в 5 дней, в последующем — раз в 10 дней, а до 150 дня введения — 2 раза в год.
Больным проводится подавление ингибиторов иммуносупрессивными препаратами (Преднизолон, Азатиоприн или Мабтера). Для остановки кровотечений применяются препараты шунтирующего действия: